Using DNA sequencing in your Genes in Space proposal
Guest post by Kiana Mohajeri
Kiana will be available to discuss this post and answer your questions about Genes in Space at the next #GenesInSpaceChat on Tuesday, April 10th at 8pm EDT. Submit your questions for Kiana HERE.
In a previous blog post Holly, one of the other Genes in Space mentors, covered PCR -- a method that creates many copies of a chosen piece of DNA. For more information on this technique check out Holly’s Facebook Live session and blog post or watch the Genes in Space PCR video. One of the main things to remember is that PCR works by making lots of DNA, in a process called amplification, but even after making billions of copies of DNA you still can’t see it. So, after PCR, we need to take advantage of methods that help us analyze that amplified DNA further.
Lots of different techniques are used after PCR to help visualize or read the amplified DNA. For your Genes in Space proposal you do not need to understand the nitty-gritty of how these techniques work. However, it can be helpful to know what information you can learn using different DNA analysis techniques in order to select the best one for your proposal.
All of the Genes in Space experiments to date have relied on a DNA analysis technique called gel electrophoresis. Gel electrophoresis allows you to see the DNA you amplified by PCR and separates the molecules by size using an electric field. It's a very useful and commonly used technique. For more details check out our video.
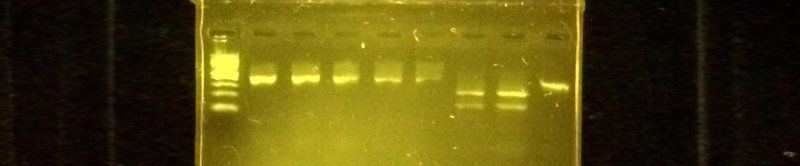
DNA visualized using gel electrophoresis.
Sophia Chen, one of the 2017 Genes in Space winners, will use capillary electrophoresis to analyze her DNA. Capillary electrophoresis uses similar principles as gel electrophoresis but allows us to determine the size of the DNA molecules much more precisely. Using capillary electrophoresis we can differentiate between molecules that differ in length by as little as 1 DNA base. If you are interested in learning more about the details of capillary electrophoresis check out this (very detailed) paper.
Gel and capillary electrophoresis are very important techniques that allow us to detect DNA molecules and tell us about their size. Often that's all you need to know to answer your experimental question. However, electrophoresis techniques don’t tell us anything about the order of the bases within the molecule. For that we’ll need a technique called DNA sequencing. This technique was used in the Genes in Space 3 mission to help identify some of the microbes living on the ISS. Through the use of sequencing, scientists are able to learn a great deal about our genetic information, getting down to the actual nucleotide bases that make up each DNA fragment we’re interested in.

DNA being sequenced in space for Genes in Space 3.
DNA (or RNA) sequencing is not a required part of a Genes in Space proposal and it's not an appropriate read out for all DNA experiments. However, we do a lot of sequencing in our lab and it’s a method I’m especially interested in, so we’re going to focus largely on sequencing for the rest of this post. If you're interested in sequencing, read on to learn more about the nitty gritty details.
What are you sequencing? What will it tell you?
In the simplest terms, sequencing will tell you the order or "sequence" of nucleotide bases that are contained within a DNA fragment. Whether you're considering DNA or RNA, the order of the bases is tremendously important.
Let's give an example using a segment of DNA in the genome that corresponds to a gene. We know that the "sequence" of DNA in a gene determines the sequence of the RNA transcribed from it, which in turn determines the order of amino acids that make up a protein. We care a great deal about proteins because they're basically the machinery of a cell -- if some task needs to be carried out, chances are it's going to be done by a protein. Certain mutations in the DNA sequence of a gene can result in a completely different protein than was originally encoded by that gene-- the protein might gain a function it wasn't originally supposed to have, lose the ability to carry out what it was originally supposed to do, or not actually form a protein at all. Although we can't study proteins with PCR, we can study the DNA or RNA that was the original blueprint for a protein. One way you can think about PCR is an indirect way to look at a protein of interest by taking a look at its blueprint.
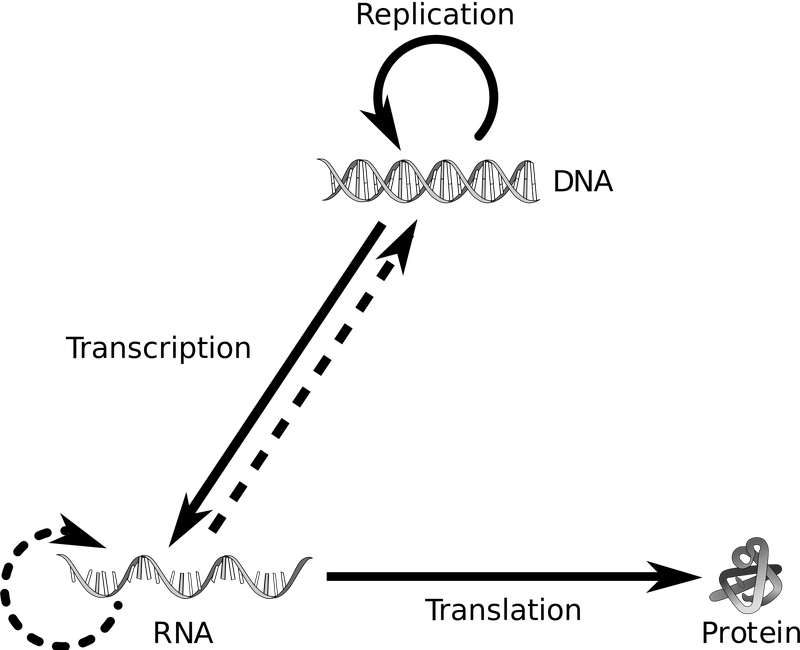
We know that the sequence of DNA in a gene is important because it can directly impact the function of the protein it encodes. However, we also know that only thinking about DNA in terms of genes is pretty over-simplified. Roughly 98% of the genome is "non-coding", meaning it doesn't follow the model of being transcribed and translated into protein. Yes, 98% of the genome of each cell does not contain a gene--then what's in those areas? For many years, scientists considered these "non-coding" areas to be "junk DNA" but we now know that is far from the case, they are actually very important.
Many of these non-coding areas of the genome are known to act indirectly on genes and influence their expression (again, indirectly affecting how much of each protein is produced in the cell). Certain sequences can serve as "DNA landing pads" (which we call motifs) for different proteins to bind. The protein won't recognize the DNA and know it can land there if the sequence isn't "just so". Different protein binding motifs are scattered all around the genome and are responsible for helping to turn on genes, turn off genes, or re-organize how DNA is folded in the nucleus (which can also have a major impact on, you guessed it, proteins). Therefore, knowing the sequence of DNA bases at non-coding sites is also important to understanding how proteins are functioning.
The major takeaways
for this section are:
1. By using PCR-based methods, we're looking at the sequence of DNA and RNA.
2. DNA and RNA sequence is really important -- they're often the
blueprint for proteins.
3. There's more to the genome than just genes. A large part
of the genome is actually responsible for regulating genes and determining
when/how they're expressed into proteins.
4. Whether we're studying genes or non-coding space that
regulates genes, many of the questions scientists are asking either directly or
indirectly get at "how is a protein affected by a sequence
change?"
How does sequencing work?
Over the years scientists have developed many methods to read the sequencing of a DNA molecule. In order to use sequencing in your Genes in Space proposal, it's not necessary to understand the details of how these techniques work. However, if you are curious, here are a few of them:
Sanger sequencing
Let's say you've done a PCR and you want to know what the sequence of your PCR product is. The simplest type of sequencing you could do is called Sanger sequencing -- the simplest type because it will tell you the sequence of only 1 PCR product at a time.
Sanger sequencing is great if you have just a few small regions of DNA that you're interested in. Some examples for when you might want to use Sanger sequencing would be if you want to check an organism to see if a certain gene contains a mutation or if you want to confirm the identity of a certain PCR product. You'd first amplify a DNA target by PCR, then Sanger sequence the PCR product to read the string of nucleotides that make it, and then you could align the sequence to a DNA database. This would allow you to confirm that your PCR had indeed amplified the target you were looking for and you could compare the sequence of your PCR product to the consensus sequence present in the database to see if there were any differences which might be due to mutation.
So how does Sanger sequencing work?
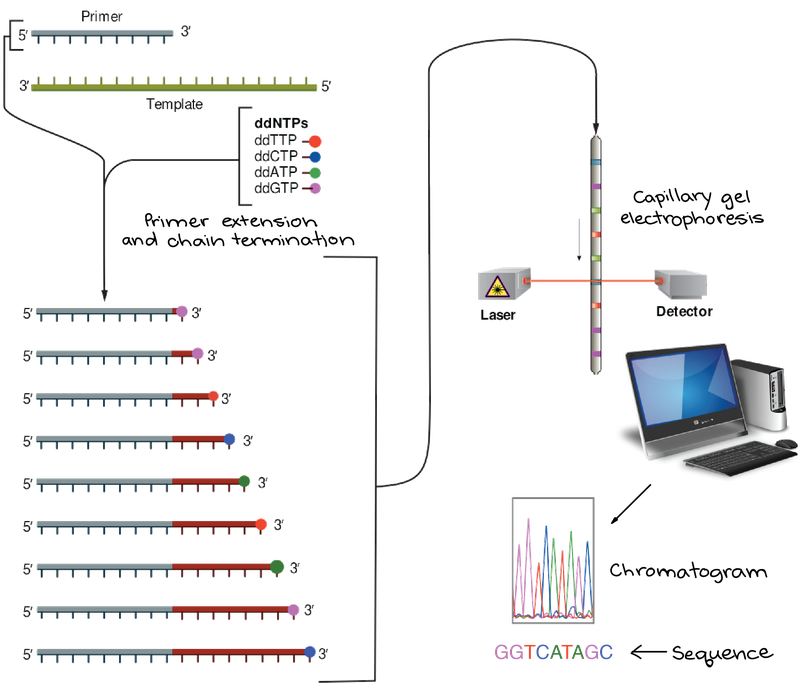
Sanger sequencing overview
To conduct Sanger sequencing you PCR amplify your region of interest and then you put it in a reaction with one of the primers you used to amplify the fragment in the first place (only the forward or the reverse) mixed with a special polymerase and two types of free nucleic acids. Type 1 (dNTPs) are ATGCs that can have additional nucleotides added to their 3’ end, and type 2 (ddNTPs) are ATGCs that are terminal and cannot have additional bases added to their 3' end. Nowadays they also usually have a fluorophore dye on them, a different color for each of the four nucleotide bases.
Similar to PCR, the polymerase uses your PCR
product as a template to build more DNA molecules. As this polymerase is making
more copies of each DNA molecule, it’s using the free bases you’ve added to the
mix, usually adding a type 1 base but every now and then, adding a type 2 base. If
a type 2 base is used it stops the polymerase because it cannot add another
nucleotide to the 3’ end of these bases. Given that you have many molecules of
your PCR product floating around, just by random chance you'll see these type 2
or terminal bases incorporated at different positions of your PCR product. This
will give you many fragments of DNA of different sizes because the polymerase
stopped copying at different places along the molecule. Different sequencing or
gel-based methods are able to separate out DNA fragments that differ in size by
a single base, allowing a visualization of which terminal base was incorporated
at each position of the original PCR product, telling you the sequence of the
PCR product.
Want to learn more? Check out this website, this animation, and this video.
Sanger sequencing is one of the older DNA sequencing techniques but it is still useful – I submitted some samples for Sanger sequencing myself a few days ago. The drawback with Sanger sequencing is that it only allows you to sequence a few hundred bases at a time. When you have 1-20 sites you're interested in sequencing, Sanger is probably the best technique to use. However, the human genome is 3 billion letters long. It would be very tedious to sequence the entire genome a few hundred letters at a time. In fact, the Human Genome Project sequenced the entire human genome and relied on Sanger sequencing. It took hundreds of scientists over a decade to complete!
Luckily, we now have new sequencing technologies that allow you to pool together many PCR products in a single sequencing reaction. These are called high throughput sequencing methods.
High throughput sequencing
The major difference between Sanger and high throughput sequencing is the number of DNA fragments able to be sequenced at once. With high throughput methods you can determine the sequence of 100s to 1000s of DNA molecules in a single shot. This drastically increases efficiency and makes these methods great for sequencing multiple sites of DNA at once. You could very rapidly determine the sequence of the whole genome if that's what your experimental question requires.
There are a few different classes of high throughput sequencing but the two major ones we'll focus on are:
1) Next Generation sequencing -- "NextGen"
2) Third Generation sequencing -- "ThirdGen"
The main differences between NextGen and ThirdGen sequencing is the chemistry behind them and the length of the DNA molecules they're able to sequence. NextGen sequencing uses some of the same techniques as Sanger sequencing but you can read many DNA molecules at once. To learn more about how NextGen sequencing works, check out this video and this article.
Similar to Sanger sequencing, NextGen techniques can only sequence DNA molecules that are a few hundred base pairs in length. The last step in the NextGen sequencing process is to take all of the short sequences and using a computer to assemble them and map them back to known sequences of the genome (thanks to the Human Genome Project, we know what the sequences of each chromosome should look like). This is useful for a lot of applications, but if certain sequences are repeated many times in the genome it can be difficult to map them using NextGen sequencing.
ThirdGen sequencers fill a different important
niche because they are able to sequence long pieces of DNA, as many as 60,000 bases from a single DNA molecule. There are several different methods including nanopore sequencing which is used on the International Space Station. Check out this video for more about sequencing on ISS.

Astronaut Kate Rubins sequencing DNA aboard the International Space Station for the first time.
We've covered multiple types of sequencing, from the older, lower throughput technology of Sanger sequencing to the newer NextGen sequencing and the cutting edge ThirdGen sequencers. I want to end with mentioning that you really should not get bogged down by how cool or new a technology is when designing an experiment. It really comes down to the question you want to ask. Some questions are better suited to targeted approaches (like Sanger sequencing) while others require a broader approach that benefits from high throughput or longer reads. More info isn't always better. Keep your research question and hypothesis in mind in your experimental design -- if the hypothesis would benefit from a higher throughput approach, great! If not, there are tons of scientific questions out there that need to be answered and can be approached with what some may consider "old school" methods.

